Structural Dynamics of Ion Channels and Transporters
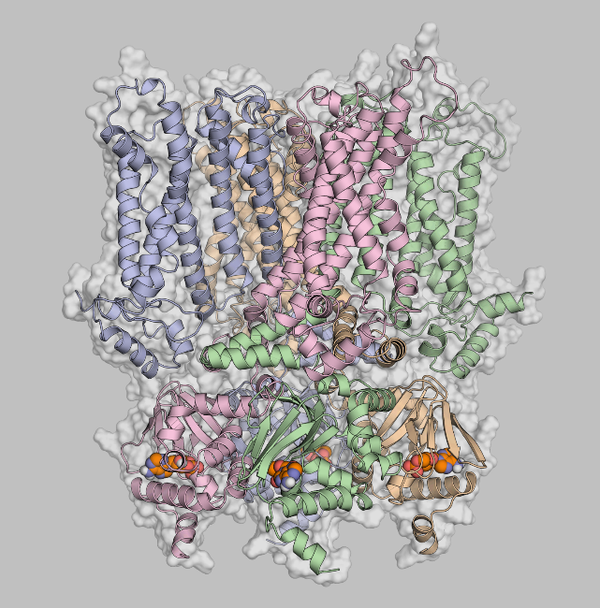
Ion channels and transporters are present in the membranes of all living cells and mediate a variety of cell functions. Ion channels enable the rapid change in membrane permeability for ions such as sodium, potassium, and calcium. As a result, they generate and modulate electrical signals in cells. Ion transporters adjust the concentration of ions inside and outside of cells. As biomolecules that are heavily involved in basic life processes, ion channels also play an important role in understanding disease mechanisms and therapeutic approaches.
Recently, the German Research Foundation has extended the funding of the research group FOR 2518 “Functional Dynamics of Ion Channels and Transporters – DynIon”, which has been investigating the functioning of these passage gates through the cell membrane for three years. The computational chemist Prof. Holger Gohlke (Institute for Pharmaceutical and Medicinal Chemistry of the Heinrich Heine University Düsseldorf; NIC research group Computational Biophysical Chemistry at JSC, Forschungszentrum Jülich) and his team will investigate hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channels, dysfunction of which may give rise to diseases. At the atomistic level, the scientists want to use molecular simulations and modelling to understand how the channels are activated by way of coupling membrane potential and ligand binding. Molecular dynamics simulations will be performed in this context on the GPU partition of JUWELS. These studies will be complemented by experimental data from the lab of Prof. Klaus Benndorf from the Friedrich Schiller University Jena.
Contact: Prof. Holger Gohlke,
h.gohlke@fz-juelich.de
from JSC News No. 274, 30 July 2020