NIC Excellence Project 2017/2
Ab-initio-Untersuchung von Grenzflächen: Eigenschaften und Reaktionen

John von Neumann Exzellenzprojekt 2017;
Prof. Dr. Michael Moseler, Fraunhofer Institute for Mechanics of Materials, Freiburg
Im bisherigen Verlauf des Projekts „Ab-initio-Untersuchung von Grenzflächen: Eigenschaften und Reaktionen“ konzentrierten sich die Untersuchungen mittels quantenchemischer Simulationen auf drängende Fragestellungen der Energiewende. Die Simulationsaktivitäten erstreckten sich dabei von einem tieferen Verständnis und letztendlich einer Kontrolle der elektronischen Struktur von Halbleitermaterialien, Halbleitergrenzschichten und Halbleiter-Elektrolyt-Übergängen zur Unterstützung der Entwicklung photoelektrochemischer Zellen für die solare Wasserstoffproduktion über phononische Eigenschaften neuartiger „hot carrier“ Solarzellen-Materialien bis hin zu katalytischen Eigenschaften von Materialien und Oberflächen, die für neue Syntheserouten für die Treibstoffproduktion aus CO2 eingesetzt werden können.
Im weiteren Projektverlauf soll die Untersuchung von Halbleitermaterialien und deren Grenzflächen für die Verwendung als Elektroden zur Wasserspaltung weitergeführt werden, wobei nun in Kollaboration mit Prof. Sanjay Mathur von der Universität Köln auch außergewöhnliche Materialien wie Uranoxid in Augenschein genommen werden sollen. Dies mag auf den ersten Blick seltsam erscheinen. Es steht jedoch eine enorme Menge an schwach-radioaktivem Uran als Abfall aus der Nuklearwirtschaft bereit, dessen Lagerung große Probleme bereitet. Eine Wiederverwendung dieses Materials in einem sinnvollen Kontext ist daher äußerst erstrebenswert. Die wissenschaftlichen Fragestellungen sind dabei die Verwendbarkeit und Kompatibilität von Uranoxid in Heterostrukturen mit anderen Oxiden für die Anwendung als Photoelektrode zur Wasserspaltung sowie die Kontrollierbarkeit der Lage der Bandkanten bei solchen Heterostrukturen.
Zudem soll abermals im Feld der Tribologie geforscht werden, indem tribo-chemische Reaktionen von Umgebungsgasen mit Oberflächen und Poren von amorphem Kohlenstoff untersucht werden. Hierdurch sollen Einblicke in Reibungs- und Verschleiß-Mechanismen von Diamant- und Diamant-artigen Beschichtungen gewonnen werden, um das hervorragende Reibungs- und Verschleiß-Verhalten solcher Kohlenstoffhartschichten aus atomistischer Sicht zu verstehen. Amorpher Kohlenstoff bildet sich als Zwischenphase bei der Kollision von Rauigkeits-Spitzen aus. Dessen Wechselwirkung mit der Umgebung im Reibspalt ist daher von essentieller Bedeutung für das Erlangen eines solchen Verständnisses. Weiterhin soll ein weiterer Schritt in Richtung eines grundlegenden Verständnisses von Trockenreibung erlangt werden, indem Wasserstoff-, Fluor- und gemischt-terminierte Diamantoberflächen als wohldefinierte prototypische Systeme untersucht werden. Die Modellsysteme sind unkompliziert und lassen sich mittels klassischer Kraftfelder beschreiben. Quantenchemische Rechnungen sind aufgrund der Beschränkungen der Zeit- und Raumskalen für die geplanten Scher-Simulationen nicht praktikabel. Zudem lassen sich die Kraftfeldparameter wie Atomladungen und elastische Eigenschaften unabhängig voneinander variieren und so deren Einfluss auf das Reibverhalten studieren. Letztendlich könnten daraus Regeln zur gezielten Materialmodifikation abgeleitet werden, um ein gewünschtes Reibverhalten einzustellen.
Die beschriebenen Forschungsaktivitäten fußen auf großskaligen numerischen Simulationen auf dem Rechner JURECA. Für die Beschreibung der elektronischen Struktur an Grenzflächen sowie von chemischen Reaktionen an solchen finden quantenmechanische Ab-initio-Methoden (Dichtefunktionaltheorie) Verwendung. Für Simulationen, die größere Raum- und Zeitskalen erfordern, wird Molekulardynamik mit klassischen Kraftfeldern eingesetzt.
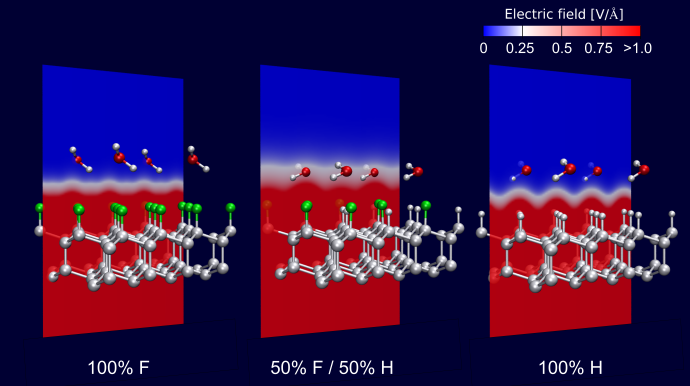
Abb. 1: Auf den zu 100% mit Fluor bzw. 100% mit Wasserstoff terminierten Diamantoberflächen (links und rechts) fällt das elektrische Feld sehr rasch ab, so dass Wassermoleküle im praktisch feldfreien Raum (blau) adsorbiert werden. Auf der gemischt mit Wasserstoff und Fluor terminierten Oberfläche (Mitte) ist das elektrische Feld ausgedehnter und Wassermoleküle können in Bereichen hoher Feldstärke (weiß, rot) adsorbiert werden.