NIC Excellence Project 2021/2
Bridging drastically coarse-grained and microsopic descriptions in hierarchical modeling of Soft Matter. Application: Non-linear viscoelasticity of polymer melts

John von Neumann Excellence Project 2021
Prof. Kurt Kremer, Max Planck Institute for Polymer Research, Mainz, Germany
Polymers are chain-like molecules consisting of thousands of similar monomers linked by chemical bonds. This thread-like molecular architecture renders high-molecular-weight polymer liquids non-Newtonian, with long relaxation times. Currently, the viscoelastic behavior of polymers for small deformations is well understood within the classical reptation theory and its refinements. The key idea is that the dynamics of long polymer molecules is controlled by temporal topological constraints, the entanglements. The latter arise from local non-crossability of chains, caused by microscopic excluded volume, and long-range correlations related to chain connectivity. The average number of monomers between two consecutive entanglements is defined as the entanglement length, Ne. Entanglements force a test chain to “reptate” along the contour of an effective tube which, by itself, is equivalent to a random-walk-like trajectory in the three-dimensional (3D) space. The slow reptation along the contour of this highly curvilinear 3D trajectory leads to protracted chain dynamics. Yet, modern reptation theories have limited use for strong deformations where fundamental mechanisms of polymer relaxation are still under debate. We emphasize that the regime of non-linear rheology presents significant interest not only for fundamental polymer physics but also for industrial processing.
Our goal is to unveil molecular-level mechanisms underpinning non-linear viscoelasticity using molecular-based simulations which emulate rheological experiments. Such simulations require as an input equilibrated samples of high-molecular-weight polymer melts. However, the protracted polymer dynamics precludes the preparation of such samples with conventional methods of Molecular Dynamics (MD) simulations. In this project, we achieved a breakthrough by developing a novel hierarchical backmapping strategy which can equilibrate melts with thousands of polymers and degrees of polymerization that are by orders of magnitude larger than Ne. As illustrated in Figure 1, melts are first efficiently equilibrated on mesoscopic level, using a model where polymers are described by chains of soft spheres. Each sphere represents a sub-chain with a large number of microscopic repeat units. The initial mesoscopic configuration is gradually fine-grained into representations of higher resolution until microscopic details can be reinserted using short MD simulations. The method is applicable to both: generic bead-spring and chemistry-specific, all-atom, melts. For example, we have applied our approach to equilibrate polystyrene. This basic commodity polymer is well suited for testing modeling methods because it has sufficiently complex molecular structure with features such as tacticity and bulky units (benzene rings).
We use such equilibrated melts of bead-spring polymers as an input for generic rheological experiments based on massively parallelized MD simulations. We first focused on the regime of linear rheological response and verified basic predictions of reptation theory for high-molecular-weight polymers for reference. Currently, we are investigating a broad spectrum of open questions related to the regime of non-linear rheology and the glass transition. For example, we have recently addressed the intriguing question of how entanglements evolve during polymer relaxation after strong deformation. The computational time granted by the John von Neumann Institute for Computing, enabled us to probe the relevant relaxation times, which are far beyond time scales that are typically studied in the literature.
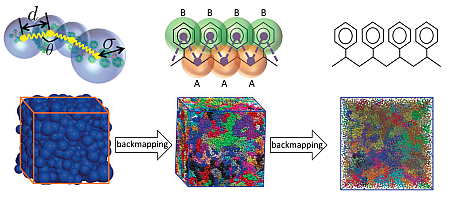
Figure 1
Copyright: Prof. Kurt Kremer, Max-Planck-Institut für Polymerforschung, Mainz
Paper selection:
Hsu, H.-P.; Kremer, K.: Efficient equilibration of confined and free-standing films of highly entangled polymer melts. The Journal of Chemical Physics 153 (14), 144902 (2020)
Hsu, H.-P.; Kremer, K.: Clustering of Entanglement Points in Highly Strained Polymer Melts. Macromolecules 52 (17), S. 6756 - 6772 (2019)
Zhang, GJ; Chazirakis, A; Harmandaris, VA; Stuehn, T; Daoulas, KC; Kremer, K: Hierarchical modelling of polystyrene melts: from soft blobs to atomistic resolution
SOFT MATTER 15 (2), 289-302 (2019)
Hsu, H.-P.; Kremer, K.: Chain Retraction in Highly Entangled Stretched Polymer Melts. Physical Review Letters 121 (16), 167801 (2018)