Force fields for redox reactions
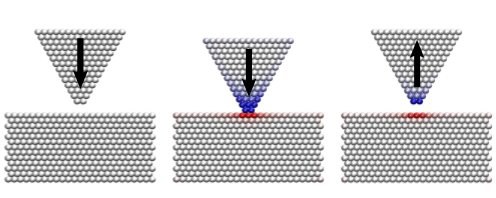
Force-field based simulations allow one to gain atomic-scale information on the chemical and physical properties of molecules, clusters, and bulk materials. The usual assumption we make when designing potential energy functions describing the interactions between atoms is that the forces between atoms are defined if we know all atomic coordinates. This, however, is insufficient to describe redox reactions during which the transfer of electrons from one atom to another atom occurs: after such a transition (i.e., in quantum mechanical terminology, after hopping from one Landau Zener level to another one) atoms have barely moved in space but dramatically altered their interactions. To account for such changes, we introduced the well-known phenomenological concept of oxidation numbers into classical force-field based molecular dynamics simulations. Forces, partial charges, etc., then depend not only on atomic coordinates but also on the oxidation numbers. This allows for the description of charge transfer within molecular systems, as sketched in the charge transfer between a dielectric tip and a dielectric substrate.
Reference:
W. Dapp and M. H. Müser, Towards time-dependent, non-equilibrium charge-transfer force fields
Eur. Phys. J B 86, 337 (2013)
DOI: 10.1140/epjb/e2013-40047-x